Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Çocukluk Çağının Semptomatik Nöbetleri
Ruhsal Durum Muayenesi
EPİLEPSİ
CH 15: Metal Şekillendirmenin Temelleri
Siroz komplikasyonlarına yaklaşım
Çocukluğun Epileptik Ensefalopatileri
İnme Sonrası Erken Nöbet Gelişimi
Türkiye Multipl Skleroz Derneği Ana Tüzüğü PDF formatında olup
Premenopozal ve postmenopozal kadınlarda lipit
PDF
Malignite ile ilişkili genodermatozlar
Ekim 2014
10. Çekirdek Dışı Kalıtım.pptx
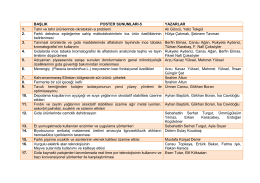
BAŞLIK POSTER SUNUMLARI-5 YAZARLAR 1. Tahıl ve tahıl
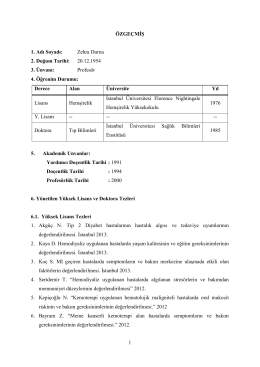
Prof. Dr. Zehra DURNA - İstanbul Bilim Üniversitesi
acil çocuk hastanın değerlendirilmesi-2014
Prematürite, postmatürite
Dillerin Sınıflandırılması
i. Plevral hastalık
PSİKOLOJİ BİRİMİ MART 2015 ÇALIŞMA PROGRAMI
1. c v x Y Yukarıda verilen venöz dalga trasesi aşağıdaki
2014/Kasım Heber Bülteni..
Çocukluk Çağı Epilepsilerinde Cerrahi Tedavi İlkeleri