Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Odabrane oblasti primenjene hemije 1 UVOD U INDUSTRIJSKU
bioprocesno inženjerstvo
biologija
Program - Tehnološki fakultet
Knjiga izvoda - EnviroChem 2013
DETIA DEGESCH PRIRUČNIK Za sigurno rukovanje i
T.C. GAZİOSMANPAŞA ÜNİVERSİTESİ
TG – 1 - İhtiyaç Yayıncılık
HAYAT BOYU ÖĞRENME KURUM
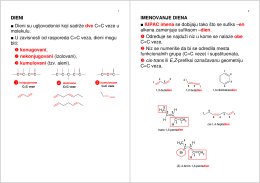
DIENI Dieni su ugljovodonici koji sadrže dve C=C veze u molekulu
Alkeni
Protect those you love with PNA life insurance Protect those you
Enzimi - Biolozi 2011/12
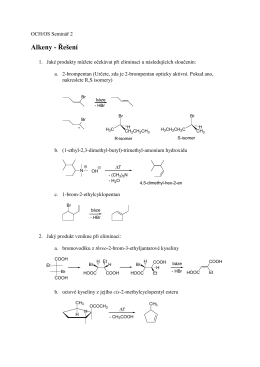
Alkeny - Řešení
pdf,1000KB - Tehnološko
GEOMETRIJA ORGANSKIH MOLEKULA
pdf,1305KB - Tehnološko-metalurški fakultet
NSAIL ANALGOANTIPIRETICI
НАСТАВНО-НАУЧНО ВЕЋЕ П О З И В - Tehnološko
Wykaz dziedzin nauki i technik według klasyfikacji OECD
afiša - Univerzitet u Tuzli
ecb 2016 - Klaster LifeScience Kraków
flexibilného rozsahu - Štátny veterinárny a potravinový ústav