Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
HematoLog - Türk Hematoloji Derneği
mezenkimal kök hücre klinik uygulamaları
HematoLog - Türk Hematoloji Derneği
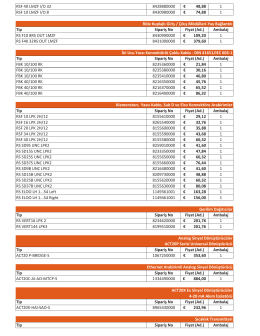
RSF 40 LMZF I/O 32 8428880000 48,88 € 1 RSF 10 LMZF I/O 8
Midenin Burkitt Lenfoma Olgusu ve Literatürün
Edinsel Bağışıklık_2
Dermatolojik Acil Durumlar
7-Göz Yaşartıcı Gazlar Ve Deri Bulguları. Dr. Mualla Polat
HematoLog - Türk Hematoloji Derneği
2014-2015 öğretim yılı bahar dönemi türk dili ve edebiyatı bölümü
HematoLog - Türk Hematoloji Derneği
erişkin orak hücre hastaları için transplant protokolü geliştirme
Bedensel Hastalıklarda Görülen Ruhsal
Uz. Dr. Atike Gökçen Demiray
INŽENJERSKOGEOLOŠKI I GEOTEHNIČKI ISTRAŽNI RADOVI ZA
Białaczka włochatokomórkowa i inne rzadsze postacie białaczek
KLL`de Yeni Ajanların Kullanımı
Tedaviye Dirençli Epilepsi Olgusunda Asistol Sonucu
Paroksismal Nokturnal Hemoglobinüri Tanı ve Tedavi
Halkın Seçeceği İlk Cumhurbaşkanının Kişisel Bağışlarla
KAN
İÇİNDEKİLER - Türk Hematoloji Derneği
Lenfadenopatili ve Splenomegalili Hastaya Yaklaşım