Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Bolum 048 - ResearchGate
Bolum 019 - ResearchGate
POSTOPERATİF ADJUVAN KEMORADYOTERAPİ UYGULANAN
Difüzyon Ağırlıklı MR Görüntülemenin Pankreasa Ait
PDF Fulltext - Gaziantep Medical Journal
teknik şartname
EYLÜL 2014 DÖNEMİ 3. DENEME SINAVI TEMEL BİLİMLER SORU
Ders5
Bölüm I - endoteam - Süleyman Demirel Üniversitesi
Kit ve Kit Karşılığı Cihaz temini alımı
77 - İstanbul TTM
Sayı 17 - İzmir Yüksek Teknoloji Enstitüsü
Rukset Attar
Jinekolojik Kanserlerde Tarama
Türkiye Kanserle Savaş Vakfı
KOLOREKTAL KANSERLER
Kranyal ve spinal vasküler hastalıklarda radyocerrahi Sunum planı
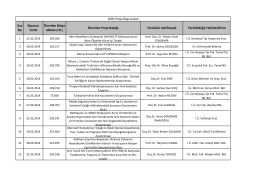
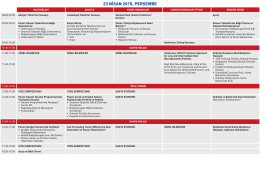
23 NİSAN 2015, PERŞEMBE
LOKAL İLERİ EVRE PROSTAT KANSERİNDE HORMONAL TEDAVİ
" pdf " dokument
tc istanbul bilim üniversitesi sağlık bilimleri enstitüsü histoloji ve
Glioblastoma tümörlerinde çoklu ilaç direnci