Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Amino Acid Degradation
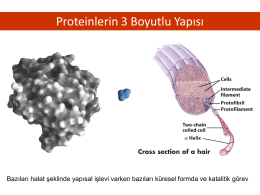
Proteinlerin 3 Boyutlu Yapısı
Sermet Sağol
Amino Asitler, Peptitler, Proteinler
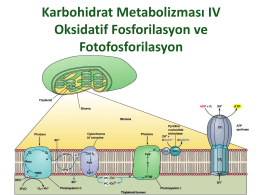
Karbohidrat Metabolizması IV Oksidatif Fosforilasyon ve
B6 Vitamini
Gülistan Bahat Öztürk
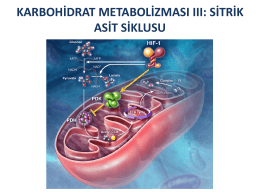
KARBOHİDRAT METABOLİZMASI III: SİTRİK ASİT SİKLUSU
AMİNO ASİT METABOLİZMASI - Doç. Dr. Ali Vaiz Garipoğlu
ÖZET (PDF) - Akuademi.Net
Deneme PDF dosyası
Detaylı Özgeçmiş - gıda mühendisliği
Elektrokoagülasyon İle Endüstriyel Atıksu Arıtımı
Amino asitlerin hücre içindeki reaksiyonları
Amino asitlerdeki azotun akıbeti-üre döngüsü
Pazarlama Araştırmalarında Deneysel Yaklaşımlar
Tam Makale/PDF
Amino asitlerin sınıflandırılması
Elif Damla ARISAN İletişim Bilgileri
Специальное предложение Термальный курорт
Amino asitlerin sınıflandırılması
emniyet yönetim sistemi el kitabı
Toxicopathological evaluation of Picralima nitida seed aqueous