Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Úvod do proteomiky
Rozhovor: Brigitte Sattmann
Bioprospect_1_11.qxd:Layout 1
Separace směsi proteinů na zařízení pro izoelektrickou fokusaci v
témata v rámci projektu BOX - FBMI
Serie 1 - řešení - Studiumbiologie.cz
Ukázka z knihy Genesis konflikt Když se podíváme na
Effenberková Lenka_Jak využít arteterapii a artefiletiku v hodinach
Abstrakty příspěvků
13. listopadu 2014 / November 12
Günümüz genomik çağında modern tıp
Tübitak projesinde çalışmak üzere burslu Yüksek Lisans (Tezli)
Rámcová témata disertačních prací pro akademický rok 2015/2016
Atomová a jaderná fyzika - Modularizace a modernizace studijního
PROGRAM - Elektro 2014
CECE 2010 program
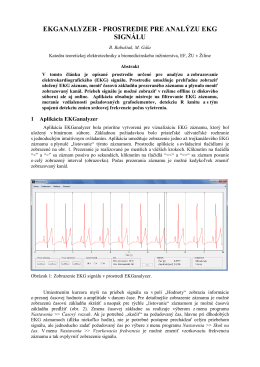
EKGANALYZER - PROSTREDIE PRE ANALÝZU EKG SIGNÁLU
Hmotnostní spektrometrie Mass Spectrometry (MS)
Návrh a konštrukcia lekárskych prístrojov
Adopce a barrandovský Videostop PDF