Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
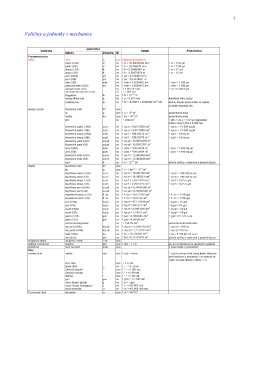
soubor ke stažení - Modularizace a modernizace studijního
1.Základné poznatky o molekulách Ciel`om je zopakovat` základné
1 ÚVOD
stáhnout - Gymnázium INTEGRA BRNO
Optické vlastnosti koloidních soustav
James Clerk Maxwell a zrození dynamické teorie
Část I Termodynamika
Sbírka úloh z mechaniky (autorem je doc. Habrman)
vybrané otázky
prezentace - Fyzika GJVJ
tttropaplyn - Střední průmyslová škola kamenická a sochařská
2-Stavové chování
Abstrakty příspěvků