Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Štatistická fyzika
1.Základné poznatky o molekulách Ciel`om je zopakovat` základné
Vybrané kapitoly z kvantovej fyziky
3.7.4 Trecie sily Celkom všeobecne, trecie sily pôsobia proti smeru
Vyhl`adávanie v texte
Informačný materiál
Změna teploty při osolení vody a ledu
ŽILINSKÁ UNIVERZITA V ŽILINE DIPLOMOVÁ PRÁCA
FX [f:ks]
Predikátová logika
Ako hrať - eCasino
Procesy, 1. ˇcast` - osa.dcs.elf.stuba.sk
Predikátová logika
Vysoká škola báňská – Technická univerzita Ostrava
AMERICAN ROULETTE 1. Pravidlá hry a. Hraciu plochu
soubor ke stažení - Modularizace a modernizace studijního
1/2013 - Transfer technológií - Centrum vedecko
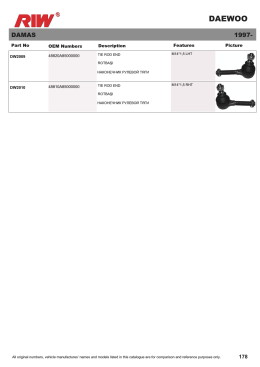
DAEWOO
(6. – 7. 12. 2012) – Call for papers
5. prednáška
dost o spolupráci
ČJ: UZSVM/BJI/7676/2015-BJIM
slajdy2011/pp12fs.pdf - osa.dcs.elf.stuba.sk







![FX [f:ks]](http://s2.readgur.com/store/data/000255313_1-2d6c11f75a52c1574d71330a9f90f6f1-260x520.png)