Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
FYZIKÁLNÍ CHEMIE I a SEMINÁŘ Z FYZIKÁLNÍ CHEMIE 2
sešit 1 - Ksicht
Opora z fyzikální chemie (inovovaný prozatímní učební
zde - Ústav petrologie a strukturní geologie
Část I Termodynamika
přednáška ke stažení, pdf, 375 kB - Ceramics
soubor ke stažení - Modularizace a modernizace studijního
Unterstützt durch: Politika územní spolupráce se týká prostoru EU
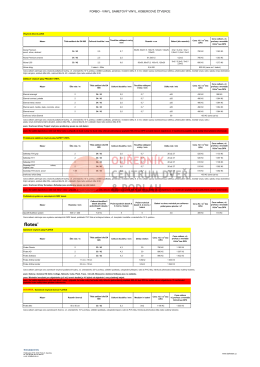
FORBO - VINYL, SAMETOVÝ VINYL, KOBERCOVÉ ČTVERCE
Návod k obsluze TRAMEC_stručný - MOTOR
Vyziva_remake_Rencin - Prof. MUDr. Karel Martiník, DrSc.
HYDRAGEL ISO-CK K20
Optické vlastnosti koloidních soustav
Katalog větracích systémů Lossnay
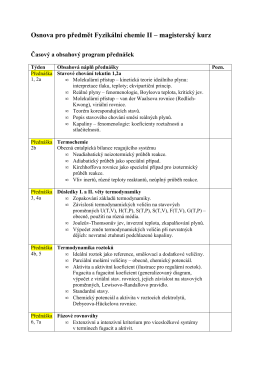
Sylabus prednášky a cvičení
Příklady
PDF Střednědobá revize Víceletého finančního rámce
Mekanik Şartname
fázové přechody
Pece a energetické hospodářství.pdf
Fridex Eko - Velvana as
Neurogenní dysfagie u dospělých: Petra Kučerová
Document