Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Tam Metin (PDF) - Türk Pediatri Arşivi
ügff*J - mut ilçe millî eğitim müdürlüğü
Liposakşın ve yağ enjeksiyonu
Ek-1 Bakım Standartları Malzeme Listesi
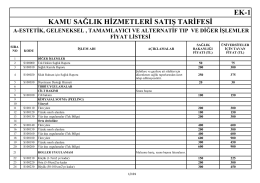
EK-1 KAMU SAĞLIK HİZMETLERİ SATIŞ TARİFESİ
Havva Öz Alkan - Türk Kardiyoloji Derneği
Prof. Dr. Fikrettin ŞAHİN
UPEK KITAP.indb - Ulusal Pediatrik Endokrinoloji ve Diyabet Kongresi
read PDF - Endokrynologia Pediatryczna
Silikon Vadisi ve Girisimcilik
PDF Olarak İndir - Perinatal Dergi
EPIGENETIKA
Erciyes Medical Journal
Kadın Genital Kadın Genital Hormonları Fizyolojisi
Boy
Erciyes Medical Journal
Volume 46 - Subject Index 46. Cilt
Projeden Elde Edilen Sonuç ve Öneriler
Yağ enjeksiyonu - Cosmedic İstanbul
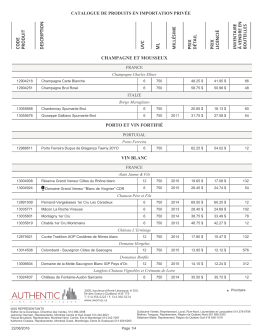
importation privée
Journal Of Turgut Ozal Medical Center
Dosya Yükle - Türk Psikiyatri Dergisi
Evrensel Eklem Simülatörü PDF