Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
00 yeni.indd - Türk Hematoloji Derneği
Kurumsal Eşitlik Endeksi 2014
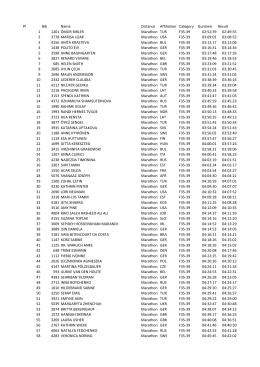
Maraton Bayanlar 35-39
Chýrnik - Poľno SME
Bu PDF dosyasını indir
HematoLog - Türk Hematoloji Derneği
Peter Dzurák - Ako sa žilo v Bošáci II.
Tarihi II” dersini DÖNEMLİK ALAN ÖĞRENCİLERİN DİKKATİNE!
Akut Lösemiler
Zarządzenie 230/2013 - Platforma PWSZ w Ciechanowie
Čti více
HEMATOPOİETİK KÖK HÜCRE KAYNAĞI Ali Ünal* *Erciyes
DEEP OSCILLATION®
Malignite ile ilişkili genodermatozlar
Młode opakowanie
aziz polat
Ülker Koçak
Ogólny Schemat Systemu Znakowania FQ50 OPIS SYSTEMU
Bilimsel Programı İndirmek İçin Tıklayınız!
Ermenistan-Türkiye Normalleşme Süreci Destek
TTOK 2014 “Sözlü Sunum Tartışması”
program - Bach-collegium Praha
Nizip Toplum Merkezi Yöneticisi İş İlanı