Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Soubor kapitol ze stránek http://www.genetika
ZDRAVÍ a NEMOC
Sborník
Obecná zoologie - KATEDRA BIOLOGIE
Prokaryotické organismy
brožura text 34. r.soč.pdf
LABORATORNÍ PŘÍRUČKA - Patomorfologická laboratoř,sro
Diagnostika rostlinných virů - Biologické centrum AV ČR, vvi
antropologie a genetika člověka - KATEDRA BIOLOGIE
Molekulární onkologie
Jak (ne)funguje imunitní systém
sp.zn.: sukls84640/2012 Příbalová informace- informace pro
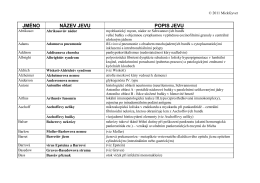
Patologický slovníček
Toto číslo si môžete stiahnút priamo tu
Vyšetření u sterility a před um.oplozením
COHN přírodní látky kolem nás
2013/2 - dermanet.eu
Fyziologie živočichů a člověka - KATEDRA BIOLOGIE
Informace pro lékaře a zdravotníky Význam - AXIS-CZ
Varilrix - Vakcíny a Očkování
Číslo dokumentu:
školící materiály k BOZP v PDF