Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
antropologie a genetika člověka - KATEDRA BIOLOGIE
Zoologické dny Brno 2015 - Ústav biologie obratlovců
Mutační analýza genů asociovaných s genetickými syndromy
Synlabianer 04/2014 - synlab Czech Republic
Vážená paní doktorko, pane doktore Laboratorní - mz
Ujíždí nám v léčbě vlak? - Národní sdružení PKU a jiných dědičných
9. výukový týden - Numerické aberace
podzim 013 - překlad 21. století
Výsledky projektu „Genetika a příjmení“ a zapojení
Soubor kapitol ze stránek http://www.genetika
Hemolytické anémie rozdělení fyziologické poznámky
Mutace, Mendelovy zákony,
vrozené vývojové vady
Dědičnost barev u BO - Klub bretaňských ohařů
stiahnuť formulár
pdf popis úloh
Spravodaj c 157 - szcpv
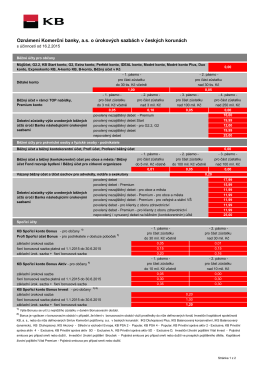
Oznámení Komerční banky, a.s. o úrokových sazbách v českých
Sylabus předmětu 5EN203 – Makroekonomie 1
Příbalová informace: informace pro pacienta Kuvan 100 mg
pro akademický rok 2014/2015 - Katedra mediálních a kulturálních
sylabus předmětu
Eugenika a ľudské práva - Prvý slovenský portál pre diplomové práce