Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
KISA ÜRÜN BİLGİSİ Bu ilaç ek izlemeye tabidir. Bu
Kısa Ürün Bilgisi
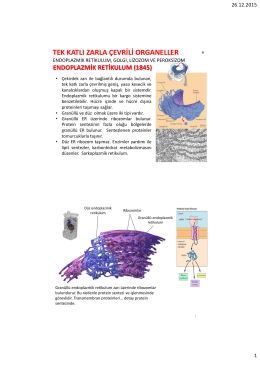
tek katlı zarla çevrili organeller
PDF formatı için tıklayınız.
Laboratuvar Yönetimi Sempozyumu Kalite
Kısa Ürün Bilgisi
KISA ÜRÜN BİLGİSİ 1. BEŞERİ TIBBİ ÜRÜNÜN ADI ADEMPAS
Etik Kurulu Çalışma Esasları için Tıklayınız.
kısa ürün bilgisi
Kısa Ürün Bilgisi
Sınav kaygısı
Document
İlaç - Sakarya Üniversitesi
Kadın Doğum Hekimi Gözüyle Sperm Parametreleri
D 6102 BD 6101 BD 6101 BS D 6082 BD 6081 B
Narkotik Olmayan Ağrı Kesiciler
Clopixol® Acuphase 50 mg/ml IM Enjektabl solüsyon
Alkol Kullanım Bozukluğunda Farmakolojik Tedavi
Foresight, Altyapıya Küresel Bir Bakış - Mart 2014 (PDF
1 /17 KISA ÜRÜN BİLGİSİ 1. BEŞERİ TIBBİ
Hazırlanacak Raporları yazım ve basımında “A4” (210x297 mm
Türkiye Cumhuriyet Merkez Bankası Olağan Genel Kurul
Selincro® 18 mg Film Kaplı Tablet