Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Işık TUĞLULAR - BE ve Faz 1 - İstanbul Üniversitesi Klinik
^^Tzrt:*^2^^»aftada dabiiecegi g,bi bazr
Formularz rejestracji na warsztaty dla dzieci 2015
PROF. DR. İSMAİL H. ULUS ÖZGEÇMİŞ (Ocak 2014) 1. Adı Soyadı
İlaç - Sakarya Üniversitesi
İstanbul Üniversitesi, Eczacılık Fakültesi İlaç AR-GE Çalışmaları
Yönerge, Kılavuzlar ve Komisyon üyeleri için tıklayınız
Araştırıcı`lar çalışma süresince temel laboratuvar kurallarına uygun
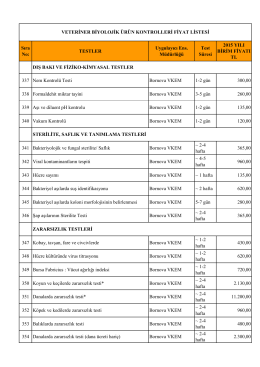
VETERİNER BİYOLOJİK ÜRÜN KONTROLLERİ FİYAT LİSTESİ için
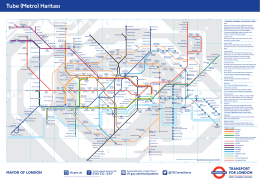
Tube (Metro) Haritası PDF 1.89MB
TDV DIA - İslam Ansiklopedisi
Kısa Ürün Bilgisi
Kısa Ürün Bilgisi
Biyoyararlanım/Biyoeşdeğerlik Çalışmalarında Olası Hata ve
Semra SARDAŞ - İstanbul Üniversitesi Klinik Araştırmalar
Ercüment KARASULU - Ege Üniversitesi
BULİMİA NERVOSA - Klinik Psikofarmakoloji Bülteni
Klinik Araştırmada Tarafların Görev ve Sorumlulukları
BİYOYARARLANIM / BİYOEŞDEĞERLİK ÇALIŞMALARI BAŞVURU
Biyoteknolojik ürünler ve Biyobenzerler Eğitim Kursu
sık sorulan sorular - Diş Hekimliği Fakültesi
Chairs: Vladimir Tesar - Ahmet Gul 08:30
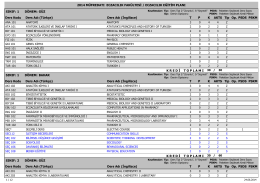
DÖNEM: GÜZ Ders Kodu Ders Adı (Türkçe)