Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
karbonhidrat metabolizması hastalıkları
Katalog aukcji - Portolan Paweł Podniesiński
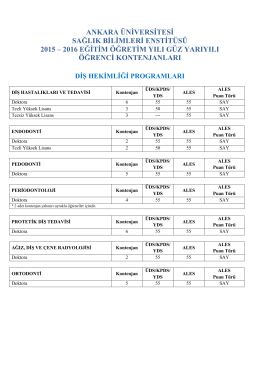
2015-16 Güz Dönemi Örgün Öğretim ve II. Öğretim
Prednáška4
küresel iş dünyasında kültürel farklılıkların yönetimi: türk girişimciliği
Postüral Drenaj Uygulama - İzmir Güney Kamu Hastaneleri Birliği
enerji
Çocukluk Çağında Siroz ve Metabolik Karaciğer
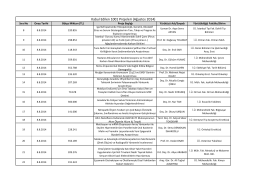
Program konferencji
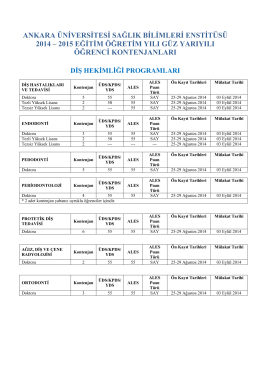
Örgün Öğretim ve II. Öğretim için Tıklayınız
Katalóg školení
Kalme_Kurumsal_TR - Kalme Kurumsal Gayrimenkul Değerleme ve
15 - İstanbul TTM
İNSÜLİN DİRENCİ NEDEN VE NASIL GELİŞİR?
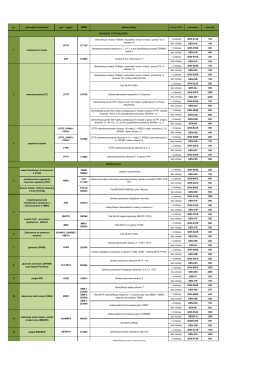
Lp. jednostka chorobowa gen / region OMIM zakres analizy izolacja
TDF/FTC - Klimik
77 - İstanbul TTM
PDF Fulltext - Gaziantep Medical Journal
Örgün Öğretim ve II. Öğretim İçin Tıklayınız
Ten szczególny dzień Ten szczególny dzień się budzi
Bolum 048 - ResearchGate
Kurşun ve Çocuk Sağlığı
Scenariusze lekcji - Kulczyk Foundation