Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Vybrané experimentálne úlohy z koloidiky - Informácie
Editoriál - Bytové družstvo, Snina
หัวข้อวิทยานิพนธ์ การทํานายความกว้างพีคในแค
Vybrané kapitoly z koloidnej chémie - Informácie
ambrotypie – mechanické poškození a přirozená degradace
FYZIK´ALNE PRAKTIKUM
p-fenolsulfonát zinočnatý – zinková soľ kys
UDÁVAME ŠTANDARD, PODĽA KTORÉHO OSTATNÝCH
3.LINEÁRNE ANTÉNY Lineárnymi anténami rozumieme také antény
zakladni typy lepidel
Prístroje na meranie fyzikálnych veličín
Názov vysokej školy, názov fakulty: Univerzita P. J. Šafárika v
Reologie 2015.pdf
PLATELIA™ Aspergillus IgG 62783 96 TESTOV - Bio-Rad
napisali o nas 3 web.pdf
VIETE SI PORADIŤ S CHRÍPKOU? - Magazín Zdravia
Optické vlastnosti koloidních soustav
Dřevohliníková okna - Dřevěná okna a dveře na míru
DECORFILM - Barvy Adex
Stiahnuť súbor
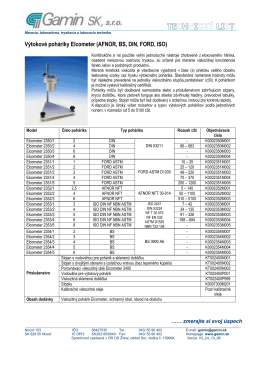
Výtokové pohárky_TL_V2_24_10_08
Koncept odborného vzdělávání
31.1 PRESNÉ VÁHY