Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
The importance of senescence in ionizing radiation
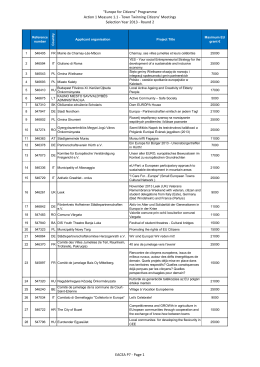
Town Twinning Citizens` Meetings Selection Year - EACEA
vždy čerstvé suroviny poctivá pena šťavnaté mäsko
univerzita mateja bela v banskej bystrici
Toplantılar Ardından
Reducing the Air Temperature Inside the Simple Structure
Studium genetické predispozice ke vzniku karcinomu prsu
Aging and cancer: molecular facts and awareness for Turkey
Valproik asit kavernozal sinir hasarı oluşturulmuş
CYTOKINETICS
Süt Sığırlarında Mevsimsel Beslemenin Sütün Karotenoid İçeriğine
Zborník prednášok - Nadácia výskum rakoviny
wykorzystanie nowego modelu doskonalenia