Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Lysosomy a lysosomální onemocnění
ZASTOSOWANIE LASERÓW
Prednáška4
Makale PDF - Güncel Gastroenteroloji
Document
Virus Epsteina a Barrové
Využití GMO ve farmacii
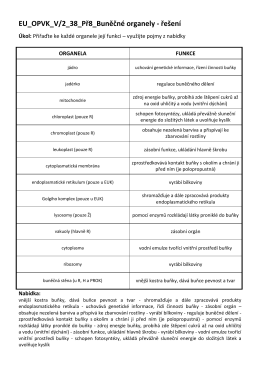
řešení
pozvánka
ZDE
Danonova nemoc (deficit lysosomálního membránového proteinu 2
Ulotka
doc. Pharm.Dr. K. Vávrová Ph.D. - Role ceramidů v kůži a
N° 20 -Trimestriel Septembre 2016 Sommaire L`agenda
sešit 5 - Ksicht
Fluorescenční spektroskopie Zadání úlohy: 1) Seznamte se s
DOPORUČENÉ POSTUPY - Klinická onkologie
Biologická léčba - Asociace inovativního farmaceutického průmyslu
LABORATORNÍ PŘÍRUČKA GENLABS s.r.o.
Atopický ekzém - Ústav imunologie a alergologie
Gaucher Hastalığı: Erişkin Hematoloğu neden
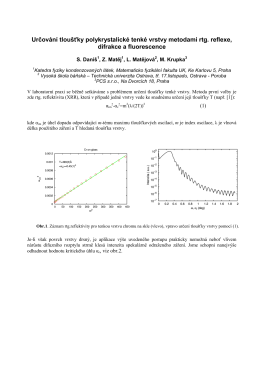
Určování tloušťky tenkých vrstev pomocí XRD, XRF a XRR
XL. májové hepatologické dny Karlovy Vary, 16