Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
Full Text PDF
`~l.JnT~Unfl`H:m1eJ
Coatings, Plastics & Polymers
Nanomateriály Nanomateriály a kapilární a kapilární elektroforéza
výzva k podání nabídky na zakázku malého rozsahu s názvem
Datasheet Usługi serwisowe
střední přirozené deformační odpory při tváření ocelí za tepla
ASİMPTOTİK GENİŞLEMEYEN DÖNÜŞÜMLER VE SABİT NOKTA
güç tutuşurluk özellik kazandırmada silikon alkoksitlerle pamuklu
“KUćA SUNCA” “OCEAN EMERAlD” “MADAM ClICQUOT”
CNC PROGRAMOVÁNÍ - FRÉZOVÁNÍ
Ağ Temelleri Hafta II
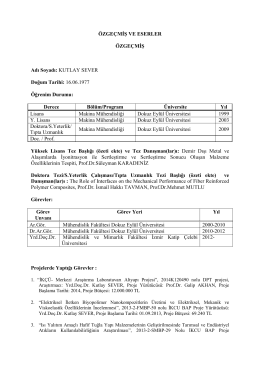
CV_Yrd.Dr.Kutlay Sever nisan2014