Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
Comparative Chloroplast Genomics Reveals the Evolution of
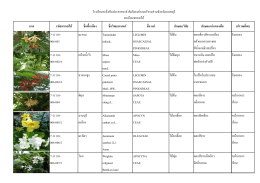
ภาพ รหัสพรรณไม้ ชื่อพื้นเมือง ชื่อวิทยาศาสตร
19. Kvartér Sborník abstrakt
gelir hareketliliği eşitsizlikleri azaltabilir mi? türkiye örneği
commercial divers as updated July 27, 2016
Two New Species of Pachytriton from Anhui and Guangxi, China
Robust Regression and Posterior Predictive
lan süs bitkileri-3 - Kocaeli Üniversitesi
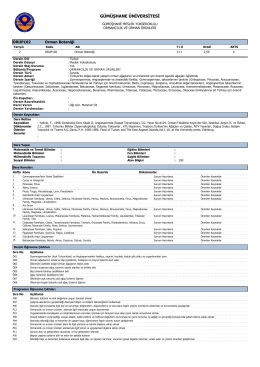
ORUP 102 Orman Botaniği - Gümüşhane Meslek Yüksekokulu
Doğu Mazısı : Geçmişten Günümüze
Liquidambar orientalis Miller - Batı Karadeniz Ormancılık Araştırma
Proje hazırlama-1
10. Çekirdek Dışı Kalıtım.pptx