Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
Dating Phylogenies with Sequentially Sampled Tips
Starodávné skládánie - Collegium pro arte antiqua
issue 15 copy - Radyo Merhaba
Sborník
TDV DIA - İslam Ansiklopedisi
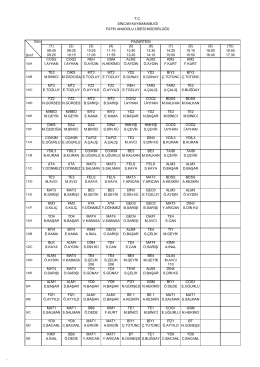
09.02.2015 Pazartesi Sınıf Ders Çizelgesi
Rotavirus enfeksiyonu - Türkiye Halk Sağlığı Kurumu
DERS 1: Sistematik Biyoloji - Taksonomi Yaz Okulu
İnsan Kaldırma Hareketinin Analizi için Tip−2 Bulanık
Robust Regression and Posterior Predictive
PDF - Wiley Online Library
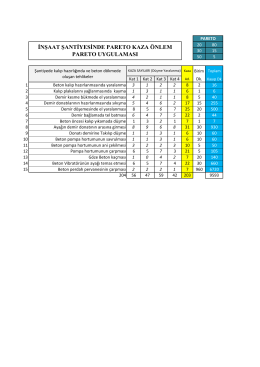
Pareto Liste
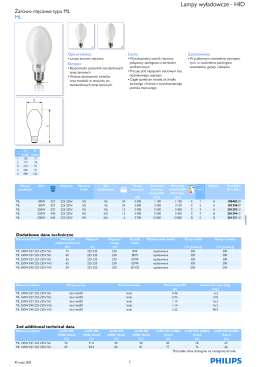
Product data sheet: ML