Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
Cloning, purification, and characterization of a thermophilic
program kitapçığı - ekoloji 2015 vı ulusal ekoloji sempozyumu
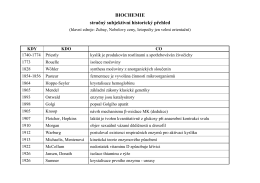
Historie biochemie
Characterization of a novel xylose isomerase
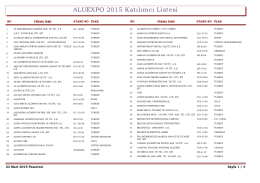
Katılımcı Listesi
Enzimlerin Genel Özellikleri, Sınıflandırılması ve
Anoxybacillus sp. 13K Bakterisinden Termofilikrabinofuranosidaz
yıldızlar ferdi sonuçlar - İstanbul İl Yüzme Temsilciliği
Cloning and expression analysis of 1-deoxy-D
ATPaz Özelliğine Sahip Olduğu VarsayılanAlüminyuma Dirençlilik
Employee shift schedule
İnguinal kanal
สารประกอบใดที่ไม่ใช่น้ำตาล