Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Robbins Patoloji Cep Yeni
จัดการบ้านหลังน้ำท่วม
Temel Tıp Bilimleri Testi
1. ‹çindekiler 2. 3. 4. 5. 6.
Serum tiroglobulin düzeyi - Türkiye Aile Hekimliği Dergisi
AKUT GÖRME KAYBI Patofizyoloji Vizyon kaybı bilateral veya
Ders - Tıp Fakültesi
histon deasetilaz inhibitörü valproik asidin endometriyum
Emas Fiyat Listesi
Ürünün içerik detayı için TIKLAYINIZ
PDF İndir - Türkiye Aile Hekimliği Dergisi
fiyat listesi 2016
Benign Meme Bulguları ve Hastalıkları
The Neuron Doctrine: Was it Just The Cell Theory
Sunum için tıklayınız.
PDF Olarak İndir - Perinatal Dergi
A.Ü.T.F. TIBBİ PATOLOJİ ANABİLİM DALI LABORATUVARLARI 2015
DUYURU
DİŞ HEKİMLİĞİNDE UZMANLIK EĞİTİMİ GİRİŞ SINAVI (DUS
PDF İndir - Türkiye Aile Hekimliği Dergisi
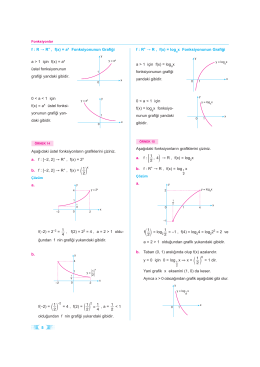
12 Matematik Eksik Konular
PDF katalog sayfası için tıklayınız
Instrukcja obsługi mantis.luji.pl