Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Postižení kardiovaskulárního systému u žen s Turnerovým
Name HCP Date of Birth Name HCP Date of Birth Lisa
abstrakt 2012:abstrakt.qxd.qxd
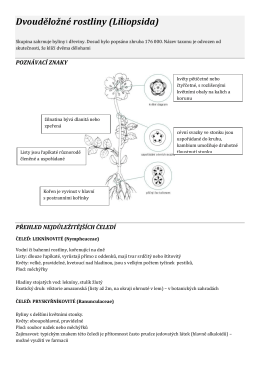
Dvouděložné rostliny (Liliopsida)
Almasa Kapidžić biografija
El-Ayak Sırtı Ödeminden Tanı: Turner Sendromu
Melanomy ve veterinární medicíně
zde - Euromedic
Adolesanda jinekolojik sorunlar ve üreme sağlığı üzerine etkileri
Kdy indikovat vyšetření srdce pomocí CT nebo magnetické
vrozené vývojové vady
Q^:=1*
Zwężenie cieśni aorty i dwupłatkowa zastawka aortalna u
Communiqué de Presse
Trizomie 18
27 Tue.
fulltext
Obez Olan ve Spor Yapan Çocukların Kardiyak Fonksiyonları ve
Protokół badania kardiologicznego u płodu w ośrodku referencyjnym
17. symposium PS Chlopenní a vrozené srdeční vady v dospělosti
zde
Jak vzniká lymfedém? - Lymfoterapie
Bábätko zomrelo po podaní 9 vakcín v jeden deň