Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
thesis - Disulfiram
Organizace, management a podpora výzkumu
Bakalářská práce pro tisk 2
T.C. GAZİOSMANPAŞA ÜNİVERSİTESİ
Nabízejí více... - Kadaňské noviny
Bulletin - Chemické listy
PDF Fulltext - Gaziantep Medical Journal
Prohlédnout - Český den proti rakovině
Buněčný cyklus a buněčná smrt
Opis zajęć - pdf - Instytut Etnologii i Antropologii Kulturowej
Česká republika
Bulletin dětské endokrinologie
informace pro uživatele VELCADE 3,5 mg prášek - Janssen
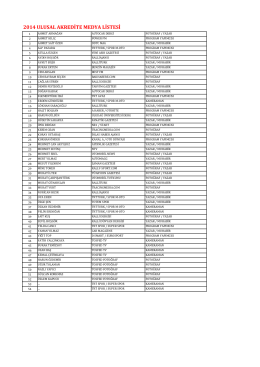
2014 ULUSAL AKREDİTE MEDYA LİSTESİ
červen 2016
Cancer chemopreventive effect of dietary Zataria multiflora
Informacje o produkcie
Časopis (pdf) - Beckman Coulter
Molekulární onkologie
Sıçanlarda Bortezomib İndüklü Karaciğer Hasarında B-1,3-(D
meme kanseri̇nde endokri̇n tedavi̇ di̇rençi̇
Bakınız
5) Fyziologie rostlinných hormonů auxinů: receptory a signální dráhy