Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
full text pdf
Karta Uczestnictwa
Miroslav Mati, Eva Matiová
OTR 1/2013 (MARZEC)
3+4/2010 - Společnost pro pojivové tkáně
Linear porokeratosis: a case report
Bütün makaleleri PDF olarak indir
Toxicopathological evaluation of Picralima nitida seed aqueous
null
5. Infekcije u hirurgiji
Elektronický dokument
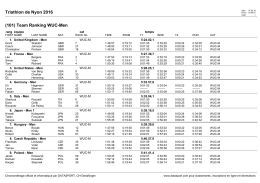
Triathlon de Nyon 2016 (101) Team Ranking WUC-Men
Servikal Vertebranın Difüz İdiyopatik İskelet