Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
Suppl 4_2008.indb
ÜRİNER SİSTEM HİSTOLOJİSİ Prof.Dr.Yusuf NERGİZ
Magazin posvecen želji muškarca za potomstvom
program festivala - Dom zdravlja Stari Grad
2011 (pdf, 3.53k)
PEDIJATRIJSKI DANI SRBIJE NIŠ 2011
Podociti glomerulska barijera Podociti, glomerulska barijera i
osobna stranica
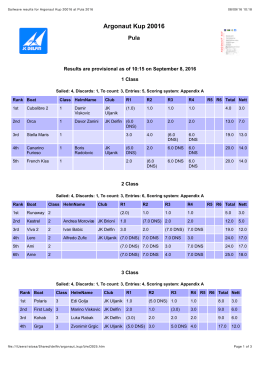
Rezultati Argonaut Kup 2016
Bir Yaş Altında Görülen Nefrotik Sendromdan Sorumlu Olan
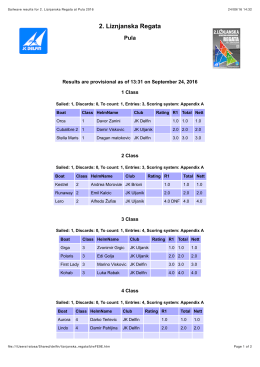
liznjan_2016_rezutati