Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Nadir Görülen Letal İskelet Displazisi: Roberts
Obezite ve Gebelik
Eylül 2014, Cilt 6, Sayı 3 - İstanbul Kanuni Sultan Süleyman EAH
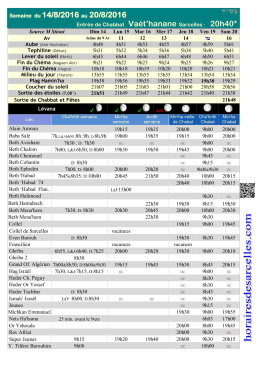
בס״ד - Horaires de Sarcelles
PDF Olarak İndir - Perinatal Dergi
Sporadik izole fokomeli
Kompletní seznam referencí
Créer un café des parents pour renforcer le lien famille/Ecole afin de
Isa Brown - ISApoultry
Hiperkalsemik hastaya yaklaþým
ROBERTS SENDROMU NEDİR? Roberts sendromu uzuv ve yüz
Vojvođanska akademija nauka i umetnosti GODIŠNJAK 2011