Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
ke stažení (pdf 8.9 MB)
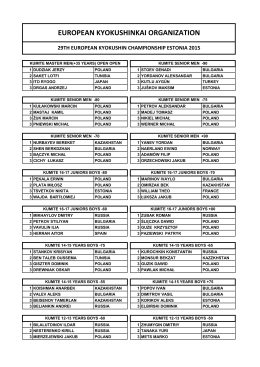
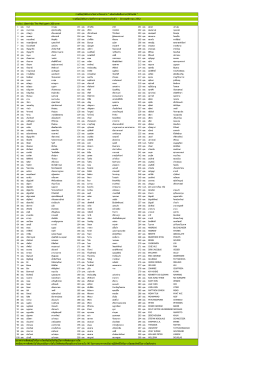
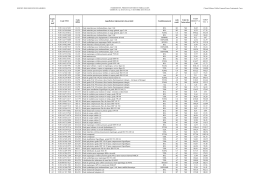
EUROPEAN KYOKUSHINKAI ORGANIZATION
Surgical treatment of achilles tendon ruptures: the
Abstracts final version (.pdf)
Optika 2.indd - Jemná mechanika a optika
Top 300 cust._Nov09
Dağıtma Makineleri
zde - Technologické centrum AV ČR
- Kopaonička škola Prirodnog Prava
PRŮVODCE - Nanotechnologie.cz
vplyv rezania laserovým lúčom na mikroštruktúru a
ZDE - Nanotechnologie.cz
TRIBUTE EXHIBITION TO Elaine Ling Abandoned
PŘÍBĚH KAPKY
PŘÍBĚH KAPKY - Akademie věd ČR
Téléchargez le programme et le bulletin d`inscription
Nejen prací živ je vědec
DRS-1908 IB POLISH
Ligne de tarif Code NNO Code IPDE Appellation réglementaire du