Read
Gur
☰
Explore Categories
Sign in
Sign up
Upload
×
Download
No category
Downloaded
HEREDITÁRNÍ NEUROPATIE
Słownik polsko-angielski mian anatomicznych dla studentów
Paradajz - Enza Zaden
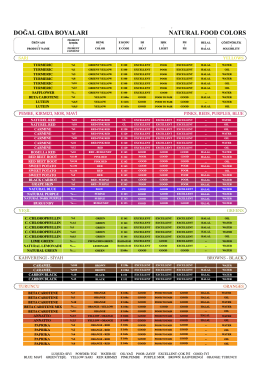
Ürün Kataloğı
Erciyes Medical Journal
Hereditární neuropatie
Žiadanka Zriedkavé ochorenia
układ kana łó w CYFR Y+
hereditárna neuropatia so sklonom k tlakovým obrnám
Modalités pour les participants français
2. Çelik Köprüler ve Yapılar Çalıştayı
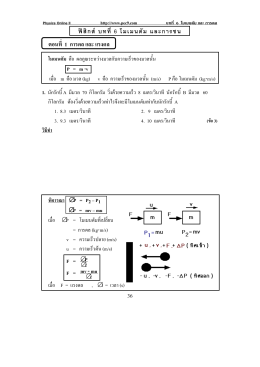
ฟ สิกส บทที่ 6 โมเมนตัม และการชน ตอนที่ 1 การดล