Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
HematoLog - Türk Hematoloji Derneği
ZOZNAM MAJITE Ľ OV / AUSSTELLERVERZEICHNIS 24.03.2012
Na‰e skúsenosti s vy‰etrovaním JAK2 mutácií pacientov s
Pengumuman Finalis PIMPI 2016
FUNKCIONALNA NOVOGODIŠNJA TRPEZA
Moleküler Onkolojik Testler ve Ci~Teknik Şartnamesi TESTİN ADI
PDF Fulltext - Gaziantep Medical Journal
P R O G R A M - OHD - Olomoucké Hematologické dny
HematoLog - Türk Hematoloji Derneği
Siyasal Parti Mensuplarının Erdoğan Algısı
8. bölüm
Biyolojik Mücadele - Mehmet Karacaoğlu Sunumu
T23_b: „Handout” of the lecture
la mission d`affaires au japon
Untitled
vox pediatriae 4/2010
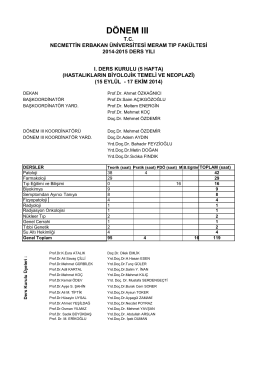
ı. kurul - Necmettin Erbakan Üniversitesi
NÁVOD K POUŽITÍ
tutaj - AdvanceMed
Syndromes myéloprolifératifs chroniques autres que la LMC
İndirmek için tıklayınız!
2014-2015 taekwondo gençler niğde grubu
CV - Groupe d`Analyse et de Théorie Economique - UMR5824