Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Mitokondri ve Mitokondri genomu
Yeni Antiepileptik İlaçlar
BİLİMSEL YÖNTEM - Biyoloji Bölümü
• 2016 LYS‐3 sınavının “ Edebiyat” bölümü sorularının müfredata
Özgeçmiş - Dokuz Eylül Üniversitesi
DEMIR EKSIKLIĞI ANEMISI
ratlarda termal injuriye bağlı oluşan gastrik mukozal hasarda
Miks Konnektif Doku Hastalığı ve Overlap Sendromlar
16. Prokaryotlarda Gen İfadesinin Düzenlenmesi.pptx
MEZANKİMAL KÖK HÜCRE BİYOLOJİSİ
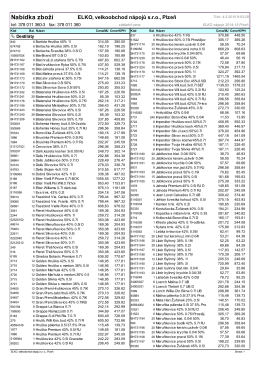
Nabídka zboží
ONPG testi - Türkiye Halk Sağlığı Kurumu
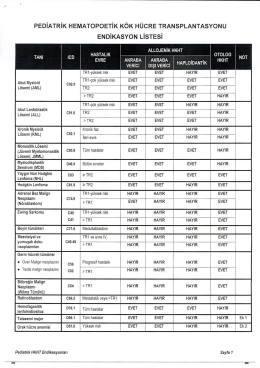
pediatrik hematopoetik kök hücre transplantasyonu
KİMYASAL ENERJİ ve HAYAT
Prof. Dr. Erbil Doğan
azalmış over rezervli olgularda oosit kalitesini etkileyen faktörler
EGZERSİZ Fizyolojisi
Adenomatöz kolon polipli hastalarda oksidatif
13. Genetik Şifre ve Transkripsiyon.pptx
π ω ω hE = ∆
görme engelli çocukların eğitim
revija 2012 br 3.cdr
05032014_cdn/adozin-3mgml-iv-enjeksiyonluk-cozelti