Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Micele jako nośniki reagentów w katalizie micelarnej
AHYDROSIL® K - Silikony Polskie
K - SSPTChem
Kongres pierwsza strona
Jeśli Twój fryzjer znów Ciebie nie zrozumiał!
Karta ECTS
program ćwiczeń
Przejdź... - Portal wiedzy
Instrukcja dla kierowców ADR
sposób wytwarzania elastycznych kompozycji epoksydowych
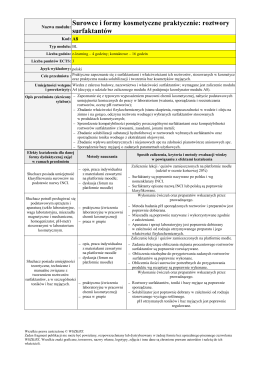
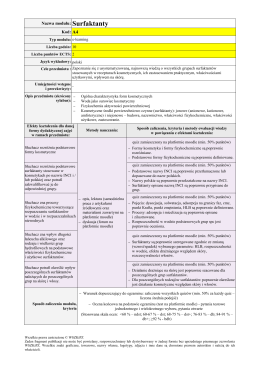
Nazwa modułu: Surfaktanty Kod: A4
Zajęcia dodatkowe - Zespół Szkół Miejskich nr 2 w Hrubieszowie
Konwersatorium - Uniwersytet Śląski
Jan Mazela Kierownik Kliniki Zakażeń Noworodka Uniwersytetu
Rozdział 3 - Zakład Technologii Chemicznej UMCS
Wyklad 2 - Leszek Niedzicki
Zespół uzależnienia od alkoholu etylowego
Regulamin konkursu dla studentów ASP w
JAK PRAWIDŁOWO WYKONAĆ DEMAKIJAŻ?
KLEJ POLIESTROWY TIKSOTROPOWY Tix +
7 spis treści
Wykrywanie GMO - Marcin Filipecki
Zwycięzcy - I Love LRP - La Roche