Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
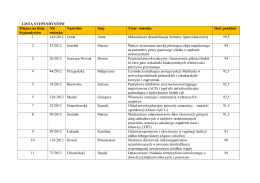
sekcje samorządu uczniowskiego - Zespół Szkół Kształtowania
Poradnik edukacyjny dla pacjentów po przebytym zawale
Print Y2772229.TIF (47 pages)
PRZEDMIOTOWY SYSTEM OCENIANIA WYCHOWANIE FIZYCZNE
echo szkoły - Szkoła Podstawowa im. prof. Józefa Kostrzewskiego w
Podstawy analizy EKG
Wykład - Miażdżyca i zawał.pdf - Diagnostyka laboratoryjna
pobierz pdf - Jakość Roku
STABILNE ZESPOŁY WIEŃCOWE
Studenci 4 semestru Inżynierii lotniczej mogą zapisać się w
Informator nr 32/2014 - Lubelskie Hospicjum dla Dzieci im. Małego
EB-A101 Jajowar (pdf)
Test Allena / Allen test/ Krzysztof Pietrzak Określenie. Badanie
15.20–15.30 Otwarcie konferencji prof. dr hab. n. med.Krzysztof
patofizjologia miażdżycy i choroby niedokrwiennej serca
ZASADY POSTĘPOWANIA W NADCIŚNIENIU
Informacja dla Pacjenta
Sprawozdanie - Projekt innowacyjne metody wykorzystania
Stop Udarom - Fundacja Udaru Mózgu
PDF file
Wybrane adipocytokiny i ich potencjalne zastosowanie w kontroli
438/PL/7 - WIPER sp. z oo