Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Návody na cvičenia z fyziológie rastlín - Informácie
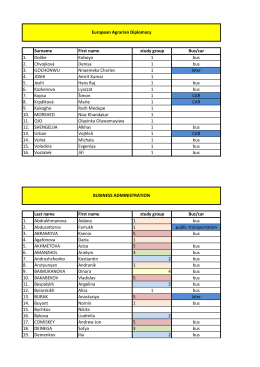
Surname First name study group Bus/car 1. Dolike Koboyo 1 bus 2
5) Fyziologie rostlinných hormonů auxinů: receptory a signální dráhy
null
kucharka_web - Centrum pedagogicko
CHÉMIA - PreSkoly.sk
Meno a priezvisko: J-1 Škola: Ekológia rias a siníc Trieda: 1. Čím je
STANOVENIE WARFARÍNU (KUMARÍNU) A BRODIFACOUMU
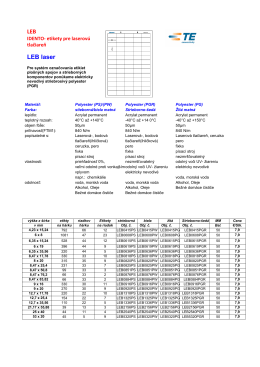
Označovacie štítky pre laserovú tlačiareň
null
null
LABORATÓRNA PRÁCA č. 1 Názov: Dôkazy prvkov v
stanovenie koncentrácie močoviny v sére, mlieku av krmive
Snímka 1
Grantové pravidlá Nadácie Volkswagen Slovakia
Oporný múr Da Vinci®