Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
DERMATOMYOZITÍDA – PATOGENÉZA, KLINICKÝ
Na stiahnutie.
Príčiny vyššieho výskytu autoimunitných ochorení u žien
LINIOVÉ STAVBY VE VYBRANÝCH STÁTECH EU
neuromuskulárne ochorenia – súčasné možnosti
Tlačová správa - Inštitút zamestnanosti
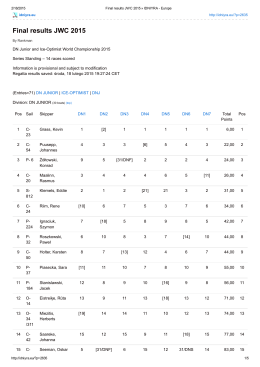
Final results JWC 2015
PROGRAM - Laboratórna medicína
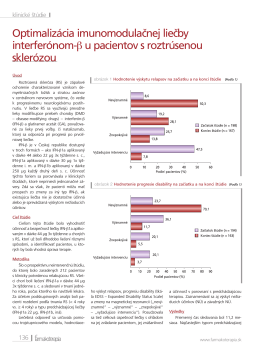
Optimalizácia imunomodulačnej liečby interferónom
Příbalová informace Forte
Akútna polymyozitída - Interní medicína pro praxi
Sborník přednášek konference OUP2012
diagnostika a liečba paraneoplastickej myasténie gravis
Tekrarlayan Periferik Fasiyal Paralizili Hastalarda Ayırıcı Tanı
správa o činnosti centra pre neuromuskulárne ochorenia
neurologické prejavy deficitu vitamínu b12 - kazuistika
makine e madrid05
1/2011 - Zákamenné
Mitochondriální DNA - Ústav dědičných metabolických poruch
Melanomy ve veterinární medicíně
Deniz Ticareti Dergisi Temmuz 2014 Sayısı
ODBORNÁ ČÁST ZÁŘÍ 2010
1 - Sekce neuromuskulárních chorob České neurologické