Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Trendy v hepatológii
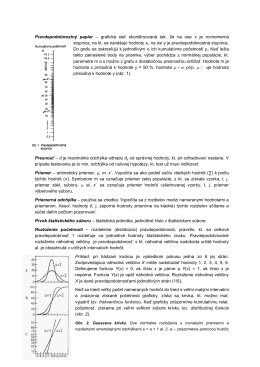
Pravdepodobnostný papier – grafická sieť
Dědičné metabolické poruchy v pediatrii
Kırmızı yansıma testi ile katarakt saptanan bir süt çocuğu olgusu
Metabo Test INFAI
12. česko-slovenská konference reprodukční gynekologie a 23
Editorial
sjait 20145-6
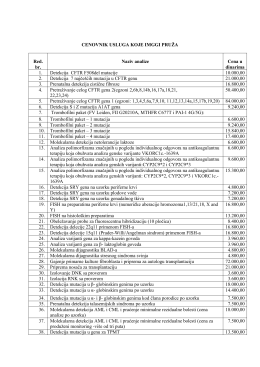
Spisak usluga sa cenovnikom
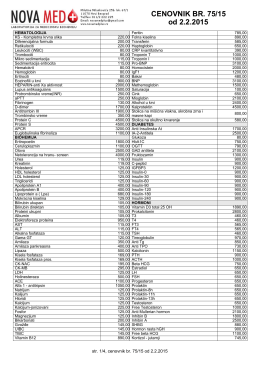
CENOVNIK BR. 75/15 od 2.2.2015
α1 – antitrypsin patří mezi skupinu plazmatických bílkovin s
Akutní jaterní porfýrie
Žiadanka Zriedkavé ochorenia
porfýrie? - sympozia.cz
ZRIEDKAVÉ OCHORENIA
Print PDF File
Print PDF File
Print PDF File
Kapitoly z “Orphanet” Encyklopédie zriedkavých chorôb
Akutní jaterní porfýrie
Pompeho choroba – liečiteľné neurosvalové ochorenie
Trendy v hepatológii
Trendy v hepatológii