Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Determination of glyphosate in groundwater - RAPAL
DERMATOLOJİK HASTALIKLARI ÖNLEME VE TEDAVİ ETMEKTE
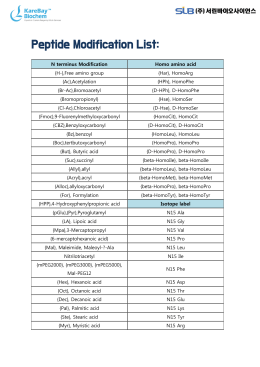
Peptide Modification List
Terminológia v analytickej chémii - Department of Analytical Chemistry
european real estate lending market
Steingrube Nakközség
Télécharger - Mathix.org
Drzwi przesuwne Architektura i design
Katalog detaliczny 2015/2016
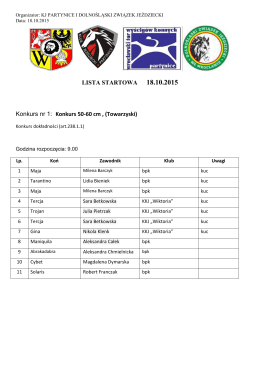
Komunikat zawodów
Télécharger mode livret
El teatro español del siglo XX en Polonia: dos
Minggu Ini - hkbp reformanda
Pobierz PDF
14 Septembre 2016 - Villes de France
Peter Ludrovský
AKCESORIA - Inter Cars
Ing. Radoslava Kanianska CSc. Publikačná činnosť
18.10.2015r. listy startowe.
Wpływ klas uformowania i otłuszczenia tusz na pH mięsa wołowego
Pobierz PDF - TAURON Biznes
İçme Sularında Arsenik Kirlenmesi
Torro - MV-servis, sro