Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Dědičné metabolické poruchy v pediatrii
LABORATORNÍ DIAGNOSTIKA Karolí a Pešková
Atopický ekzém - Ústav imunologie a alergologie
31 - Sekce klinické neuroimunologie
Ušetrete na doplatcích... (str.22)
Danonova nemoc (deficit lysosomálního membránového proteinu 2
Příloha č.4 Seznam vyšetření biochemie a hematologie
Příloha č.4 Seznam vyšetření biochemie a hematologie
zde - Imalab sro
Vršovický Zpravodaj říjen 2015
Nukleotidy jako nezbytná součást výživy pro seniory
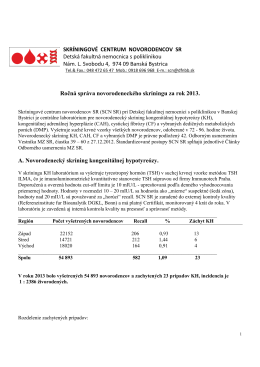
Výročná správa za rok 2013 - Detská fakultná nemocnica Banská
Dědičné poruchy metabolismu purinů, pyrimidinů a porfyrie
PDF Full text - 1. Lékařská fakulta
Trendy v hepatológii
Celý článek - Nemocnice Na Františku
Biologie - přípravný kurz 2011/2012
Zborník abstraktov z 18. Slovenskej štatistickej konferencie
PROGRAMOVÝ SBORNÍK
Výroční zpráva ÚDMP za rok 2013 - Ústav dědičných metabolických
Výroční zpráva ÚDMP za rok 2012 - Ústav dědičných metabolických
Alergenové komponenty (náhrada ISAC)
Pompeho choroba – liečiteľné neurosvalové ochorenie