Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
identification of phase composition of binders - Ceramics
1`51v1`Vl1`l
Podlahové vykurovanie - Cech podlahárov Slovenska
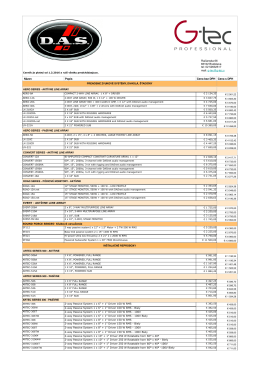
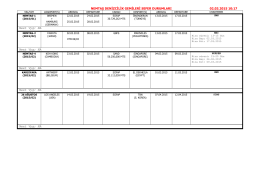
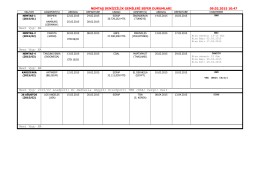
3_Cennik_DAS. Kompletný cenník - G
NEMTAŞ A.Ş. GEMİLERİ GÜNCEL SEFER DURUMU
Alkali tuz içeriğinin geopolimer cephe kaplama malzemesi
Bilten SOGFBIH br.13.. - Savez općina i gradova Federacije BiH
Drevené parkety na podlahové vykurovanie
PDF datoteka
TITLE OF THE PAPER (TNR 12p size, bold style, upper case, left
Výběr z aplikací Ramanské spektrometrie
Zprávy - Chemické listy
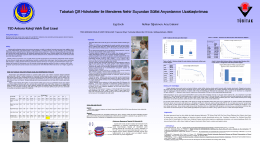
Download (1MB) - tedprints
Program skupa GNP 2012
NEMTAŞ A.Ş. GEMİLERİ GÜNCEL SEFER DURUMU
Vlastnosti betonů modifikovaných minerálními příměsmi
Základní škola Vendryně Vendryně 236 okres Frýdek – Místek www
Drevené parkety na podlahové vykurovanie
Obchodné podmienky
PRE-PORTLAND CEMENTS AND GEOPOLYMERS
1613 - İDARİ ŞARTNAME İNG. TÜRK.
25 EXPERIMENTAL CONDITIONS FOR CROSS SECTION
1718 - İdari Şartname