Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Praktikum z klasických metod analýzy
47 A skolni zadani.pdf
K POČÁTKŮM »MODERNÍHO« ČESKÉHO VĚDECKÉHO STYLU
Symetrické anténní tunery
Chemie - metodická příručka
Symetrické anténní tunery
Sborník přednášek
prozatímní učební text, srpen 2012
Elektrochemie Roztoky elektrolytů Disociace vody Disociace vody a
ARGENTI FODINA 2014 - Slovenská archeologická spoločnosť
učiteľstvo akademických predmetov, odbor Chémia v aprobácii
ELEKTROGRAVIMETRICKÉ STANOVENÍ MĚDI
PONUKOVÝ LIST
Pokročilé laboratorní metody, Advanced - EnviMod
REAKCE A VZNIK KOMPLEXNÍCH SLOUČENIN
Komplexný prospekt - Bezkontaktné izolácie
pokyny k úlohám
Vzorový výpočet - webstránke FPV UMB
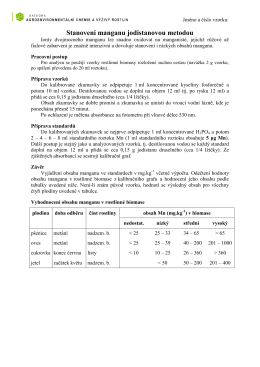
Stanovení manganu jodistanovou metodou
1108P - Welco.cz
Stanovení jodu v kuchyňské soli V analytické laboratoři je
Žádanka o laboratorní vyšetření
Predmet: ANALYTICKÁ CHÉMIA Ročník: tretí/II. polrok Forma: TEST