Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Beckwith-Wiedemann sendromu - Çocuk Sağlığı ve Hastalıkları
Full PDF - Medical Journal of Montenegro
WİLMS TÜMÖRÜ - Kanserli Çocuklara Umut Vakfı
Arşiv Kaynak Tarama Dergisi Larsen Sendromu
Vivez et Parlez Français - Özel Tevfik Fikret Okulları
[2014-11-13] 1_sunucu temel kavramlar
1218 - Ege Üniversitesi Diş Hekimliği Fakültesi
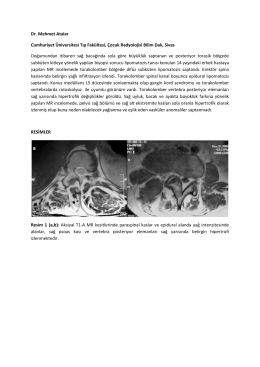
Dr. Mehmet Atalar Cumhuriyet Üniversitesi Tıp Fakültesi, Çocuk
Ders Notu 1
Kocaeli Üniversitesi Tıp Fakültesi Çocuk Sağlığı ve Hastalıkları
HematoLog - Türk Hematoloji Derneği
Ani Bebek Ölümünün Kardiyak Nedenleri
Uğur Keskin
Cavidan Gülerman
Hipertrofik kardiyomiyopati ve Costello sendromu
Prima ZOOM – příroda v dubnu 2015!
2- hakan gürkan – epigenetik modifikasyonlar ve erkek infertilite ilişkisi
Özlem Çokar - 3. Epilepsi Sempozyumu
5. ROMANTYZM
NĚKOLIK POZNÁMEK K PŘÍSPĚVKU PROF. KOTULÁNA K
Betaş Cam Mozaik
ALİ ACAR - Art Gebeliklerinde Takip Ve Problemler




![[2014-11-13] 1_sunucu temel kavramlar](http://s2.readgur.com/store/data/000019414_1-ff48adaf4f741ffa0d955ab3cdf88f4e-260x520.png)