Read
Gur
☰
Explore
Log in
Create new account
Upload
×
Download
No category
Kronik tromboembolik pulmoner hipertansiyon
Pulmoner arter hipertansiyonun genetik ve genomikleri
Pulmoner hipertansiyonda tedavi hedefleri
Sol kalp hastalıklarına bağlı pulmoner hipertansiyon
SALON 1 SALON 2 SALON 3 5 MART PERŞEMBE
Kopya Kardiyo_bahar_program_son 1.xlsx
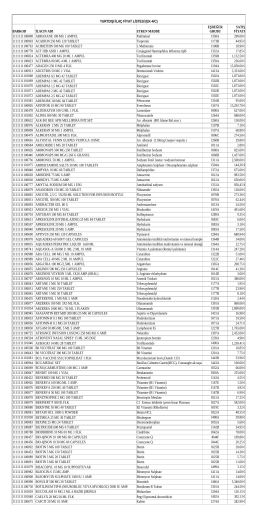
YURTDIŞI İLAÇ FİYAT LİSTESİ (EK
Pulmoner arter hipertansiyonunun güncel klinik sınıflandırılması
Pulmoner arter hipertansiyonuna sağ kalp adaptasyonu: Fizyoloji ve
Pulmoner hipertansiyon tanı ve tanımlar
1. Kurul Ders Programı - İstanbul Yeni Yüzyıl Üniversitesi
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18
Anket Cevapları için Tıklayınız.
Pulmoner hipertansiyon patoloji ve patobiyolojisi ile ilişkili konular
Kronik akciğer hastalığında pulmoner hipertansiyon
tercih edin
návod na použitie
Untitled - Türk Kardiyoloji Derneği
Kronik akciğer hastalığında pulmoner hipertansiyon
Pulmoner arter hipertansiyonunda yeni çalışma tasarımları ve
Ön ödemeli akıllı kartlı su sayacı PWM-15 PWM
Zaproszenie TaipeiPlas 2016